Next: 3 Ordalie Basics Up: ordalie_documentation Previous: 1 Obtaining and Installing Contents
A protein family can be defined as a group of proteins related through evolution that share similar 3-D structures and functions, leading usually to sequence conservation. The concept of protein family has been established in the 70's where few protein sequences and structures were known and most of them were small and constituted of a single domain. Since then, the massive increase of protein structures and sequences led to more subtle definitions, like super-family or sub-family organizations. This introduces a granulometry in the protein family concept, and gave several scales for analysis, ranging from proteins sharing only a core fold
Studying a protein family consists now in characterizing all features that specify the family, not only at a structural, functional, phylogenetic or residue conservation level, but also by using all related information available in various databases. Indeed, more and more information is available for all aspects of protein characterization, which mainly arises from high throughput technologies of the post-genomic era such as genomics, proteomics, interactomics or transcriptomics. Handling such information remains a difficult task because of its heterogeneity (3D structure, transcriptional level in time and space, ...) and deals with several levels of detail, ranging from very local data like point mutations up to large scale data like cellular localization, domain or macromolecular complexes organization or interaction. As a consequence, a new member of a protein family is then surrounded by information that can be assigned to it. Such data harvesting and assignation has been implemented in the Macsims software [10], which integrates and propagates heterogeneous information in the environment of the multiple sequence alignment of a protein family. A remaining problem resides in the analysis and the visualization of this information.
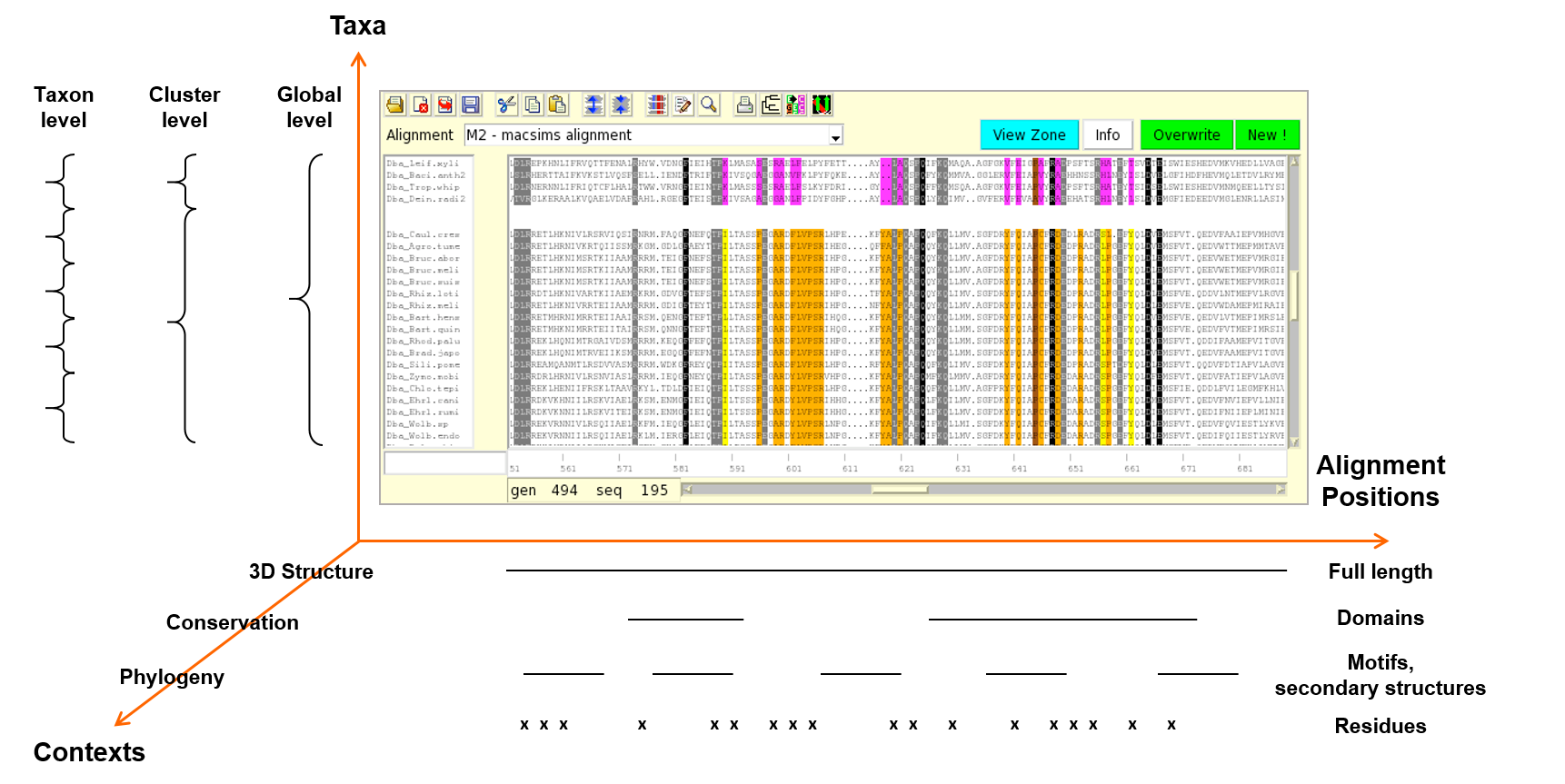
Ordalie (ORDered ALignment Information Explorer) is an interactive tool designed for the exploration of the informational content of a multiple sequence alignment into a hierarchical manner, and within different contexts, such as phylogeny or 3D structure.
The Ordalie philosophy (see fig. 1) resides in its ability to make a concomitent multi-scale analysis along three axes : the aminoacids sequence axis, the taxa axis, and the contexts axis.
The information running along the aminoacid sequence (horizontal axis) can be seen according to several scales:
Another analysis axis resides in the way the different taxa present in the alignment are handled. The study can be done at a global level (all taxa) to characterize the whole family through different features, such as conserved motifs or key signature, it can also be done on a particular taxon to identify and specify point mutation positions, or at an intermediary level to study the features allowing sub-family identification, such as differentially conserved residues between the sub-family and the other taxa.
As a third analysis axis, Ordalie embeds tools allowing different analysis contexts: residue conservation computation, phylogenetic tree computation and rendering, external features mapping, a 3D structure viewer, etc .... All analyses can be done in a structural context, as all available features can be mapped and compared on the available 3D structures present in the alignment.
For a given alignment loaded in Ordalie, it is easy to understand that many different instances of this same alignment may exist. One instance could have a given set of sequence clusters with a given sequence conservation computation, and an other instance could have an other set of clusters, in order to estimate different hypotheses. These instances are called “snapshots” in Ordalie and can be annotated, saved and retrieved at any time. This is made possible thanks to the database embedded in Ordalie.
As a conclusion of this short introduction, the strength of Ordalie for a protein family analysis resides in the cross-comparison of all information seen in different contexts and at different scales. By adjusting the coarseness of the scale (all taxa, a subgroup of taxa, or a taxon alone for example), the outcoming information will help in deciphering different aspects of the sequence - structure - function - evolution relationships for the protein family under study.