Next: 6 Menus Description Up: ordalie_documentation Previous: 4 Sequence Tools Contents
Ordalie contains a collection of tools that can be called at any time, and that can stay alive whatever happens in the main window.
The 'Snapshot Overview' is one of the available schematic representation of a snapshot. When launched, a window appears with the schematic alignment at the top and a control panel at the bottom. Any number of Overviews can be launched for a given snapshot.
In this frame, the alignment is schematized by replacing every residues by a grey pixel over a white background. It is then possible to map any feature on top of this scheme. Clicking anywhere on the scheme will automatically centre the main snapshot window on the corresponding position. On the scheme, a stippled rectangle encompasses the region shown by the main window.
Below the alignment frame, a control panel allows to interact with the scheme.
The combobox on the left is a feature selector. By default, it is set to 'automatic', meaning that all features drawn or removed in the main window will automatically be drawn or removed in the Overview window. Selecting any feature in the combobox will simply display it on the Overview.
The '+' and '-' buttons will zoom in and out the scheme.
The 'Print' button will output a PNG file of the schematic alignment in its current state. The user is prompted to give an output file name.
The 'Close' button will close the 'Overview' window.
A fruitful multiple sequence alignment exploitation ensuring a high quality of data usage by tools and feature mapping is directly dependent on the accuracy of the alignment. Although the research on algorithms dedicated to aligning sequences is still intensive and the outcoming softwares are more and more accurate, the need of manual MSA inspection, curation and editing is still necessary. This is the reason why Ordalie integrates a high performance sequence editor. It is written in C to ensure speed and fluidity, and is inspired by SeqLab [11], the editor GCG Wisconsin Package.
Entering the Editor tool will first clear the sequence from any displayed feature and colour the sequences according to physico-chemical properties. The default colouring scheme is :
(The default scheme can be changed inside the 'Edit -> Preferences' menu item).
|
|
||
At the bottom of the Ordalie window, the Control panel will display the following buttons from left to right.
Clears any current sequence selection.
This will group the selected sequences. The names of the grouped sequences will be coloured in a different colour, a unique colour for a given group. If one or more sequences already belong to a group, the user should decide through a dialog box, if the sequences should be merged with an existing group, or if a new group should be created.
Grouped sequences will behave as a single sequence.
The selected sequences will be removed from any group. If a group consists of only one sequence, the group is automatically destroyed.
By default, only gaps ('.') can be deleted or inserted, it is not allowed to insert/delete amino acids. Unlocking the sequences allows the insertion/deletion of residues.
Ordalie runs through all alignment columns and removes those containing only gaps. A 'Rem. Col. Gap.' is automatically done when leaving the 'Editor' mode.
This creates a TFA file copy of the current alignment under edition. The user is asked for a file name the first time, and successive 'Save' will use this file name to output the alignment.
Leaves the 'Editor' with all the changes made. A dialog box will asked the user if the current alignment should be overwritten, or if a new alignment should be created with the current changes.
Leaves the 'Editor' and restore the original alignment.
Clicking inside the alignment frame will show a yellow blinking cursor. The following actions are then available :
In order to speed up editing, pressing keypad or keyboard digits [0-9] stores the number in a buffer, i.e. pressing '1' then '2' will store number 12 in the buffer. Pressing then the <Left> arrow will move the cursor 12 characters to the left, and will empty the buffer. All the actions previously described can take advantage of this mechanism.
The first method consists in adding/deleting empty lines, called 'separators' hereafter, between sequence names. Separators can be added using the corresponding icon on the Icons bar, or through the 'Add separator' item of the 'Alignment' menu and deleted using the corresponding icon or item menu. The separator is added just below the selected sequence. Similarly, the separator just below the selected sequence will be removed if requested. Sequences enclosed by separators constitute then a new group. The 'Remove all separators' item of the 'Alignment' menu removes all groups in the current snapshot.
The 'Clustering' tool allows the creation of clusters (or groups) based on numerical criterions characterizing the sequences to be clustered. The computation can be done using all or part of the sequence as well as all or part of the snapshot columns. The user chooses one or more numerical criterions as the basis for the computation and a clustering algorithm. The computation can then be launched and the newly created sequence clusters are automatically displayed in the main window.
At present, the available criterions are :
Ordalie clusters and automatically defines the number of groups. The clustering algorithms along with the algorithms that define the number of clusters are taken from the Cluspack package. The available methods are :
If no sequence names are selected, the clustering will use ALL sequences. If some sequences are selected (more than 3), then the clustering will only apply to these selected sequences. The remaining ones will be kept as a separated group.
The pull-down menu allows the selection of the criterias to be used for the computation. Several criterias can be selected at the same time.
|
|
||
The 'Method' pull-down menu allows to choose the algorithm to be used for clustering computation.
The “Compute” button will launch the computation. The newly computed sequence clusters are directly displayed in the main ordalie window.
The 'Reset' button will erase any clustering done so far and show the original clustering if any. The 'No Clusters' buttons removes all groups and leaves all the sequences as a single group.
The “Clusters Names” button will pop up a window allowing to give a name to each cluster. this cluster name may be used in subsequent analysis to identify the clusters, like in the “Tree” display, or the “Barcode” tool.
The 'Save' button will leave the clustering tool and the current clustering will be saved. The user is prompted whether to overwrite the current snapshot or to create a new one. The 'Return' button leaves the Clustering tool and displays the snapshot in its original state.
The 'Tree' tool can be divided in two part. The first part consists in the tree building, which is done through the main Ordalie window. Once the tree is computed, its exploitation will be done in a dedicated new window.
The tree is computed using the FastME program using default parameters. Ordalie computes first a distance matrix based on identity percentages calculated over the selected residue range. Although Bayesian based algorithms seem to produce more accurate trees, FastME is a good compromise between speed and accuracy.
The tree can be computed on a subset of sequences and on a given residue range, for example, a region or a domain.
The left part of the Control panel deals with sequence and residue range selection.
The 'Draw' button launches the computation, and draws the resulting tree in a separate and dedicated window. The 'Return' button leaves the Tree tool.
Each newly computed tree will appear in a new and dedicated window, that allows the exploration of the tree characteristics.
Ordalie is able to render two types of trees : dendrograms and radial trees. Some of the following options are specific to one or the other tree representation (see below).
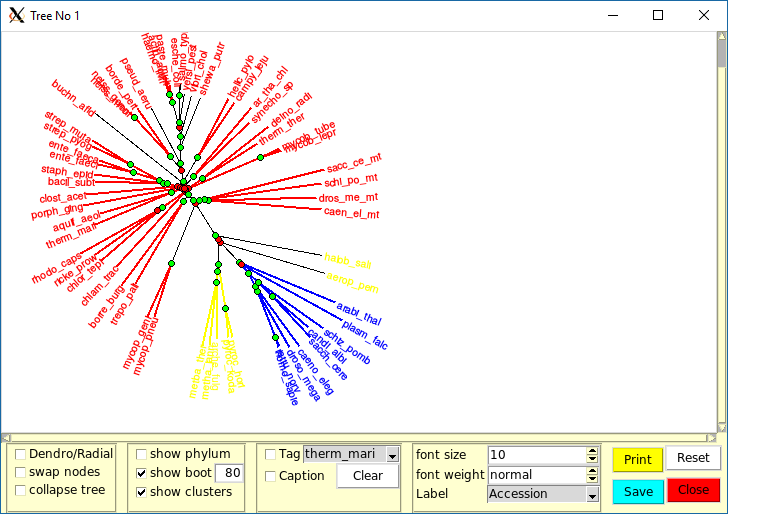 |
The upper part of the tree rendering window is the drawing area, and the bottom part the control panel area.
Adding information to the tree representation :
Tags and tree annotation :
Leaf labels :
Buttons :
Traces of the evolution pressure that maintains the structure and function of a protein family can be found while examining the residue conservation along the alignment. Both global and group conservations may help in deciphering functional sites like binding sites, interaction patches, or specialization coupled with intra-groups organization.
Ordalie offers several methods to compute conservation. Within this tool the user can try several methods to compute residue conservation. The results are temporarily kept until they are saved. A saved residue conservation computation becomes then a new feature attached to the current snapshot and can, as any other feature, be used in any tool allowing feature display.
Many methods exist to compute conservation, and they have been tested and compared extensiveley [12,4]. Ordalie implements some of them, as well as two home-made conservation methods.
The available automatic mathods are :
From left to right in the Control panel :
One of the strengths of Ordalie resides in its ability to link/map features to the 3D models (when available) of proteins. To exploit at best the feature mapping it is essential to proceed in the scope of the structural differences observed between proteins. To achieve that, Ordalie allows to superpose the strucure according to feature, and/or user defined residue range.
A protein structure can be made of several chains, which may be identical or not. A chain is usually composed of an amino acid polymer and ligands (in Ordalie, water molecules are considered as ligands). It is important here to understand that, although superposition computation are done using the polymer sequences, the entities that are moved (superposed) in Ordalie are the entire chains.
|
|
||
The chain superposition is done in three steps :
The detailed superposition algorithm is presented in Appendix 7.5.
|
|
||
Suppose the loaded alignment concerns a protein known to be a homodimer (an ![]() structure) under biological conditions, and for which several 3D structures of some proteins coming from different organism have been solved. By investigating PDB ID (say 1abc and 1def), it is also known that all structures are made of two chains, A and B.
structure) under biological conditions, and for which several 3D structures of some proteins coming from different organism have been solved. By investigating PDB ID (say 1abc and 1def), it is also known that all structures are made of two chains, A and B.
When loading the alignment, Ordalie will recognize the two PDB ID through the sequence names PDB_1abc_A and PDB_1def_B and will then download from the PDB web site the two structures with atomic coordinates, and store them inside a dedicated database. Note that Ordalie knows the coordinates for all the atoms of ALL chains of the structure, not only A. Several cases may be encountered when performing a superposition :
Ordalie doesn't know anything about monomers, dimers, multimers in general. It is up to the user to provide the information, by giving Ordalie the sequences of the chains of interest.
|
|||
If the alignment contains 'PDB\_1abc\_A', PDB_1abc_B, and PDB_1def_A, PDB_1def_B, it is then possible to superpose the two dimers. A first superposition step where only PDB_1abc_A and PDB_1def_A are selected will bring PDB_1def_A on top of PDB_1abc_A. A second superposition step where only PDB_1abc_B and PDB_1def_B are selected will bring PDB_1def_B on top of PDB_1abc_B.
The 3D Viewer is one of the most useful tool in Ordalie. Although it does not offer all the features and functionalities that would a proper Molecular Visualization program like VMD or PyMol, it can be of great help in understanding protein features in the framework of protein structures.
The Ordalie 3D Viewer is organized around the 'Molecule' and 'Object' notions. A 'Molecule' consists in all the chains (and consequently residues and atoms) that are present in a given PDB entry. An 'Object' belongs to a Molecule, and can be a composition of several elements (full chains, parts of chain, residues, ligands, etc ...) belonging to that molecule. Objects can be painted with several colours and can contain several kinds of representation. Feature mapping only applies to objects.
By default, Ordalie will create 3 objects per molecule :
Ordalie is able to represent a structure in several ways.
The 3D Viewer window can be divided in 4 parts. The top of the window is used to display information about picked atoms. Below is the 'Quick Mapping' panel. Below this panel, from left to right are the 'Molecular Objects' frame, the main 3D window, and the 'Actions' panel at the right. The 3D window can be maximized by hittin the <F1> on the keyboard, and hitting <F1> again gives the window its original geometry. All panels may be switched on or off by hitting the <F2> key.
The four comboboxes of this panel allow to make a quick mapping of features on a given molecular object. The left outmost combobox selects the molecule, then the object onto which the feature will be mapped. There are then two features selectors. It is possible to map two features on a same object by selecting one feature with 'Feature 1' combobox, and a second feature with the 'Feature 2' combobox. The features are drawn in order, feature 1 before feature 2. Care should be taken when selecting Feature 1 and Feature 2 as Feature 2 can completly cover Feature 1. For example, if Feature 1 is set to conservation, which implies residues colouring, and Feature 2 is set to PFAM-A , a lot of conservation won't be seen as a PFAM domain extends to a large range of residues. In this case, Feature 1 should be set to PFAM, and Feature 2 to conservation.
Below the 'All On' and 'All Off' buttons that switch on and off all objects that have been defined in all Molecules is the list of all 3D molecules present in the alignment. Aside the molecule name is the 'New' button that allows the definition of new objects for that molecule (see section 'Object Editor' 5.7.4 below). Clicking on a molecule name will open/close the list of the objects defined for that molecule. An object coloured in green is switched on and is displayed on the screen, a red object name means the object is switch off. Each object name is followed by the 'Edit' and 'Del' buttons, used to redefine and delete the object respectively.
This window contains the 3D objects themselves. The objects can be manipulated by the mouse through an arcball system, that is a virtual trackball. All the objects of the scene are enclosed in a sphere, and the objects are moved by dragging the sphere up and down and left to right, the mouse mimicking the hand that would roll the sphere. The mouse wheel is used to zoom in and out the scene. A right drag with <Button-3> will translate the scene in the x-y plane. A <Control-B1> click will show the label of the atom being below the mouse pointer.
Although the Ordalie 3D Viewer tool is not intended to be a complete Molecular graphics program, it still offers some functionalities which are, from top to bottom :
An object is an ensemble of residues and/or ligands belonging to one or several chains, and displayed in given styles with given colours. The Object Editor can be invoked to create a new object (the 'New' button) or to edit an existing object (the 'Edit' button).
In case of a new object creation, the new object name should be entered in the top entry box. Two objects can not have the same name.
The object edition can then be done in a five step process :
Finally, it is possible to add the molecular surface surrounding the object atoms.
When applying a color or a representation style, the user should specify the residues it should apply to. There are three ways to do so :
The object is then finished by clicking on the 'OK' button. The new object will be added to the object list on the corresponding molecule.
This representation can render several selected sequences and features on the same page. The sequences are not schematized as in the Snapshot Overview representation, but are shown as they appear in the alignment window.
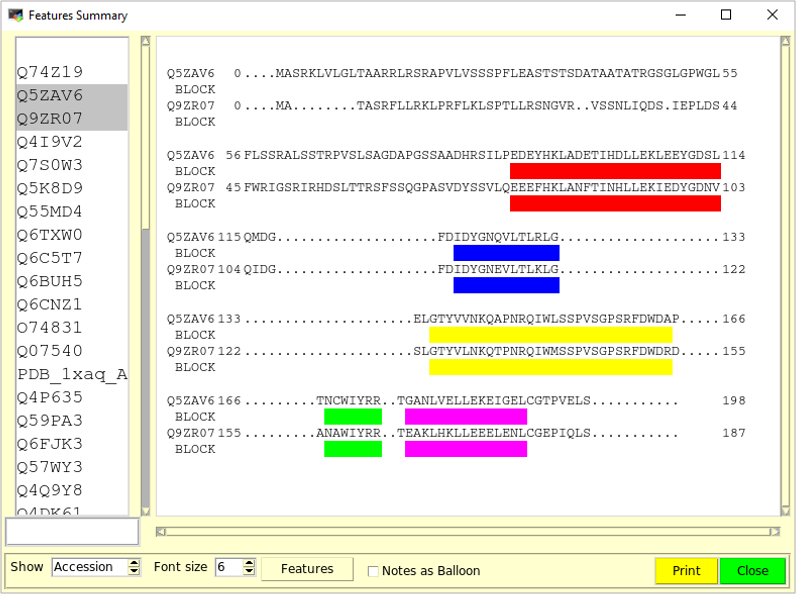 |
The top of the 'Feature Summary' window is made of a listbox containing the sequence names on the left, and a drawing area on the right. Clicking on a name selects or unselects the corresponding sequence. Multiple sequences can be selected by holding the <Control> key down while clicking on their names with the mouse.
In the drawing area, for each sequence the sequence ID is written on the left, followed by the position of the first residue in the current sequence line, the amino acid sequence itself as present in the alignment, and the position of the last residue in the line. When a feature is selected, each sequence line is followed by a feature line, with the feature name beneath the sequence ID, and rectangles below sequence positions where the feature is present.
The Feature Summary can be moved around by dragging the mouse while holding <Button-1>.
Below the Drawing Area is the Control panel. On the left is a spinbox that selects the type of name the sequence should be referenced with, i.e. its sequence name, its accession number or its bank ID, when available. This choice applies in both the listbox and the drawing area. Follows the font size selector, and then the 'Features' selector. Any number of features can be selected by checking the button corresponding to the desired feature. The 'Notes as Balloon' checkbutton renders or not the note attached to each feature as a flying balloon when the mouse pointer is over the feature. The 'Print' button will ask for a file name that will contain a PNG imageof the current drawing area, and the window will disappear by clicking the 'Close' window.
The Barcode alignment tool is an other schematic representation of the alignment.






