Abstract
The diffusion of next-generation sequencing technologies has revolutionized research and diagnosis in the field of rare Mendelian disorders, notably via whole-exome sequencing (WES). However, one of the main issues hampering achievement of a diagnosis via WES analyses is the extended list of variants of unknown significance (VUS), mostly composed of missense variants. Hence, improved solutions are needed to address the challenges of identifying potentially deleterious variants and ranking them in a prioritized short list. We present MISTIC (MISsense deleTeriousness predICtor), a new prediction tool based on an original combination of two complementary machine learning algorithms using a soft voting system that integrates 113 missense features, ranging from multi-ethnic minor allele frequencies and evolutionary conservation, to physiochemical and biochemical properties of amino acids. Our approach also uses training sets with a wide spectrum of variant profiles, including both high-confidence positive (deleterious) and negative (benign) variants. Compared to recent state-of-the-art prediction tools in various benchmark tests and independent evaluation scenarios, MISTIC exhibits the best and most consistent performance, notably with the highest AUC value (> 0.95). Importantly, MISTIC maintains its high performance in the specific case of discriminating deleterious variants from benign variants that are rare or population-specific. In a clinical context, MISTIC drastically reduces the list of VUS (<30%) and significantly improves the ranking of “causative” deleterious variants. Pre-computed MISTIC scores for all possible human missense variants are available at http://lbgi.fr/mistic.
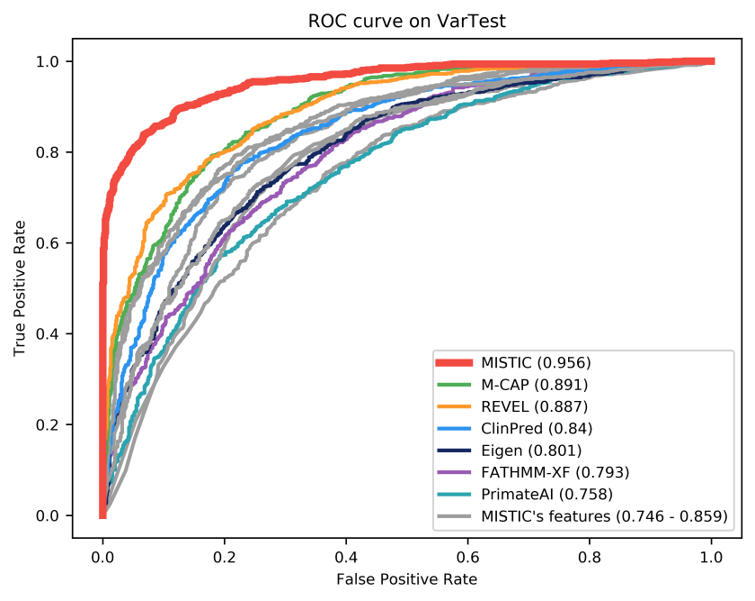
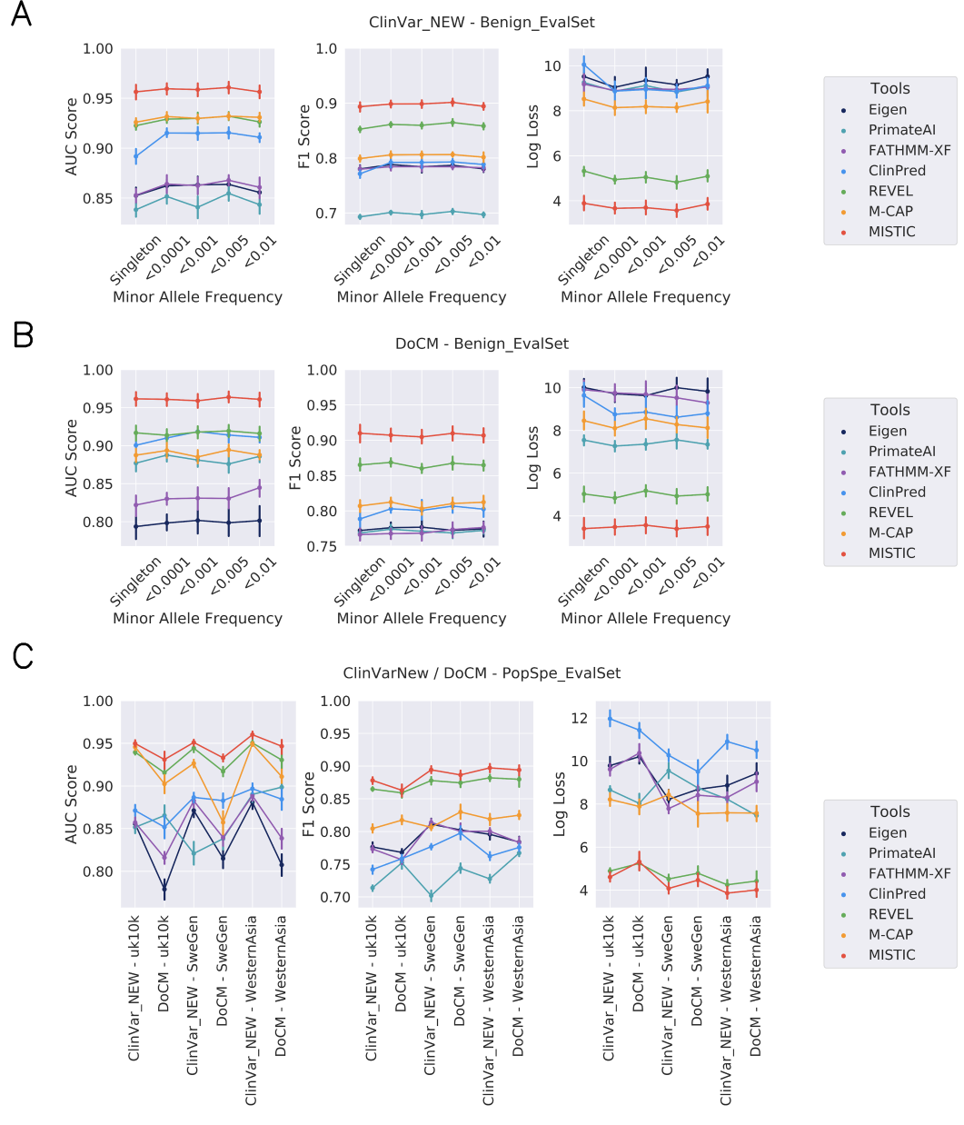
Benchmark results
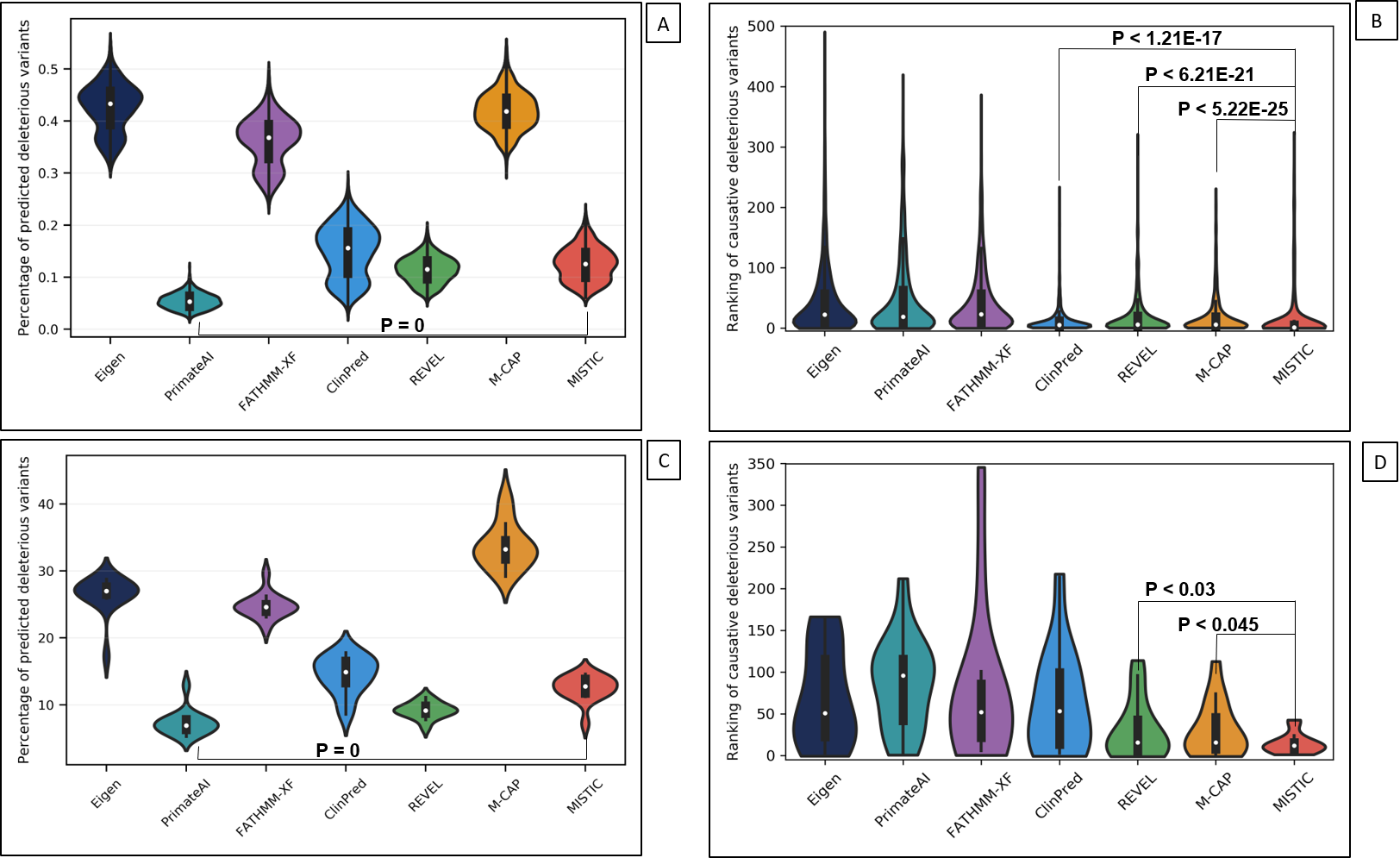
B – Ranking of the “causative” deleterious variants introduced in simulated disease exomes.
C – Distribution of the percentage of predicted deleterious variants on the exomes of the
MyoCapture project.
D – Ranking of the causative deleterious variants identified in real congenital myopathy exomes
from the MyoCapture project.
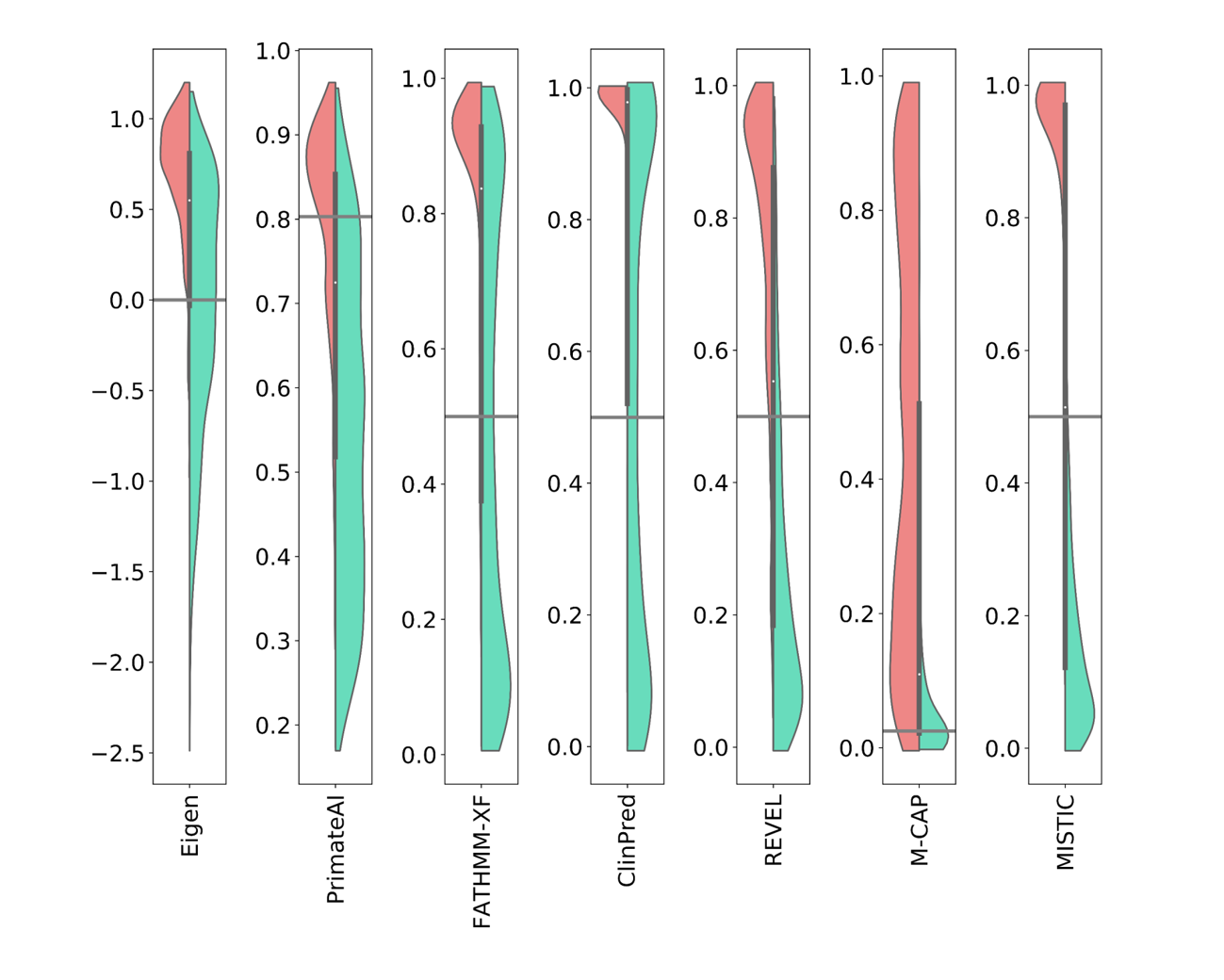
How to cite
Chennen K, Weber T, Lornage X, Kress A, Böhm J, Thompson J, Laporte J, Poch O. MISTIC: A prediction tool to reveal disease-relevant deleterious missense variants. 2020 Jul 31;15(7):e0236962. doi: https://doi.org/10.1371/journal.pone.0236962. eCollection 2020.