| Overview |
 |
 |
 |
Integrative bioinformatics for the analysis of complex systems implicated in human diseasesModern biology is characterized by huge volumes of complex, heterogeneous and noisy data resulting from i) the inherent variability of living organisms, ii) the low reproducibility of high throughput technologies and iii) the wide variety of computational and statistical analyses. This impressive flood of data means that the bioinformatics field must now evolve to isolate and filter valuable knowledge from the noise, errors or redundancy. This requires the combination of complementary automated data analysis and prediction tools with efficient storage and querying of constantly updated data. Such integrated approaches are an essential prerequisite to take full advantage of the opportunities provided by modern biology to unravel the principles and mechanisms of life and introduce them in a useful modeling of complex systems.
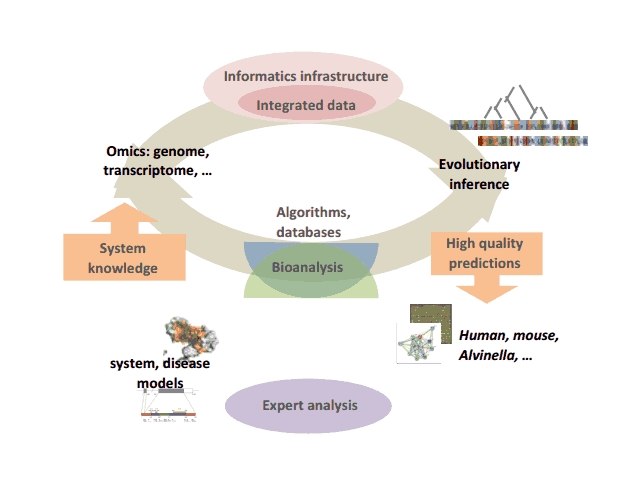
We focus on the development of robust, automated and integrated in silico approaches to study the evolution and behavior of complex biological systems (“hyperstructures”, networks, etc) in human diseases. Our major research axis concerns the exploitation of Evolution as it captures the physical, spatial, environmental and historical constraints at work in living systems and thus, if correctly deciphered, can provide a unique framework for filtering noise and for identifying shared or specific features, and subsequently propagating the inferred knowledge between biological systems. Such evolutionary inference approaches now play an important role in most high throughput studies: genome annotations, promoters, transcriptomes, proteomes or interactomes. Although many in silico tools exist, they remain largely underutilized due to the lack of a proper informatics infrastructure to make these approaches robust, accessible and easy to use in a high data flow context.
Our global strategy involves the creation of an iterative cycle combining computational developments (algorithms, databases…) to facilitate high quality human expert analysis of complex animal systems, which in turn are used to validate and refine the computational strategies.
Iterative cycle of computational development and bioanalysis
At the computational level, we are addressing several problems related to this strategy: - the design of novel algorithms, cascades of automated functional genomics data analysis, benchmark databases and expert systems for the understanding and exploitation of evolutionary information. The methodologies are developed and evaluated in the context of well dominated biological systems encompassing various levels of complexity (macromolecular structural complex, networks, diseases…) in Alvinella pompejana, mouse or human, - the development of the BIRD (Biological Integration and Retrieval of Data) system, specifically designed to automatically generate databases managing and integrating heterogeneous local or remote biological data (genomics, transcriptomics, proteomics...). BIRD provides the core for the SM2PH-db (from Structural Mutation to Pathology Phenotypes in Human-database) aimed at studying, for all human monogenic diseases, the structural impact of genetic changes in the framework of evolutionary, functional and phenotypic information. At the bioanalytical level, taking advantage of our integrated computational approaches and within the framework of longstanding international, national or local collaborations, we are participating in the analysis of complex systems implicated in human diseases. This involves notably, the study of functional impairment related to retinal or brain diseases, the identification of mutational patterns related to the Bardet-Biedl Syndrome and the characterization of genomic and transcriptomic context in various cancers. |